Pairtools walkthrough
Welcome to the pairtools walkthrough.
Pairtools is a tool for extraction of pairwise contacts out of sequencing chromosomes conformation capture data, such as Hi-C, Micro-C or MC-3C. Pairtools is used for obtaining .cool files by distiller, and has many more applications (see single-cell walkthrough or phasing walkthrough).
Here, we will cover the basic steps from raw reads to .cool file with binned contacts.
Outline:
Download raw data
“Raw” data, or .fastq files are generated by sequencing facilities or can be taken from public databases, such as SRA. We will take a sample from Rao et at al. 2017, human datasets. To reduce computateion time, take 5 mln reads instead of full sample:
[ ]:
! fastq-dump SRR13849430 --gzip --split-spot --split-3 --minSpotId 0 --maxSpotId 5000000
[1]:
! ls SRR13849430*.fastq.gz
SRR13849430_1.fastq.gz SRR13849430_2.fastq.gz
[2]:
%%bash
gzip -dc SRR13849430_1.fastq.gz | head -n 8
@SRR13849430.1 1 length=150
NTCTCAGCCTTTATAAGATAGAAGAGAGTTGGGACCTTGCTCTAAATTCTGCTTTAGCAAGGGACTTTTGTACCTGCTTTCTTCCTTTATCCAGATCTAAAAATAGTTTATATGCTGACAACTCCCTGATGTTATTCTTTGTAGTATTTG
+SRR13849430.1 1 length=150
#AAFFJJJJJJJJJJJAJAJJJJFJJJAFFFFFFA7A-FJ7JJJ-AJAJJF-<-JJFFJ7FJJF7FJJFJJ<JFAF<JJJJAJJJJA-AFJ-FJ-FF<7-A-7A-FFF<J-FJJJ<F7)A-F--7))))--77--7------A---7-7-
@SRR13849430.2 2 length=150
NAAAAGGTTTATCTCTGTGAGATGAAGCCACATAAAACCAAGCAGTTTCACAGAAACTTGTTTGTAGATTTTAAATCCGCACATCACAAAGCCATTTCACAGACAGCTTCTTTCTACTTTTTATCTGCGGCCCTTCCGTATTCCCGCATA
+SRR13849430.2 2 length=150
#AAFAJJFJJJJFAAFJA<JJJFJJFJJJJJFFJ-FF<FFJJJJJJ7AFFF<FJJ-<-F7<7<A-AF7AJAJ-A-7FF7JJJFJJFJJFFFFAJFJJ7-7-A-F--777FJFJF7---AAAF<AAA--))--))-<))-------)))7-
We next map the sequencing data to the reference genome.
Install reference genome
Although you may download and build bwa index for any genome manually, genompy can do that for you automatically:
[ ]:
# Activate bwa plugin for genomepy:
! genomepy plugin enable bwa
[ ]:
# Install hg38 genome by genomepy, it might take some time to compile the bwa index for hg38:
! genomepy install hg38
[2]:
# Check the location of the genome:
! ls ~/.local/share/genomes/hg38/
bwa-mem2 hg38_DpnII.bed hg38.fa hg38.fa.sizes index
hg38.blacklist.bed.gz hg38.DpnII.bed hg38.fa.fai hg38.gaps.bed README.txt
Map data with bwa mem
Note that you may use bwa mem2, which is x2 times faster. It proved to produce results very similar to bwa mem.
[ ]:
# Map test data:
! bwa mem -t 5 -SP ~/.local/share/genomes/hg38/index/bwa/hg38.fa SRR13849430_1.fastq.gz SRR13849430_2.fastq.gz > test.bam
After mapping, you have .sam/.bam alignment file, which cannot be interpreted as pairs directly. You need to extract contacts from it:
[2]:
%%bash
samtools view test.bam | head -n 4
SRR13849430.1 121 chr12 78795720 60 53S97M = 78795720 0 CAAATACTACAAAGAATAACATCAGGGAGTTGTCAGCATATAAACTATTTTTAGATCTGGATAAAGGAAGAAAGCAGGTACAAAAGTCCCTTGCTAAAGCAGAATTTAGAGCAAGGTCCCAACTCTCTTCTATCTTATAAAGGCTGAGAN -7-7---A------7--77--))))7--F-A)7F<JJJF-J<FFF-A7-A-7<FF-JF-JFA-AJJJJAJJJJ<FAFJ<JJFJJF7FJJF7JFFJJ-<-FJJAJA-JJJ7JF-A7AFFFFFFAJJJFJJJJAJAJJJJJJJJJJJFFAA# NM:i:1 MD:Z:96G0 MQ:i:0 AS:i:96 XS:i:21
SRR13849430.1 181 chr12 78795720 0 * = 78795720 0 TTATATGCTGACGTCGTCCATTGTTGATTCTTTGTGGTCTGTGTTCCCTCTGAAGCTCTCTGTACTTGTATATACAATTAGGCACCAACAATTCAACATGGGTAGTTATTATACTGCTAAAGCGACCCCAGAGCGAACCGGGCTACCTTN ---7---7-7)))-7---7-----7-7<---7-)<)<-----)-)<77--7---)7-<7-7----A-F<<-AF7-A777--77-7-----7--<--7-F777-A7-7---<7--7<7-<--77-77--77--7-7A---7-7---FF<A# MC:Z:53S97M MQ:i:60 AS:i:0 XS:i:0
SRR13849430.2 73 chr8 43879758 60 75M75S = 43879758 0 NAAAAGGTTTATCTCTGTGAGATGAAGCCACATAAAACCAAGCAGTTTCACAGAAACTTGTTTGTAGATTTTAAATCCGCACATCACAAAGCCATTTCACAGACAGCTTCTTTCTACTTTTTATCTGCGGCCCTTCCGTATTCCCGCATA #AAFAJJFJJJJFAAFJA<JJJFJJFJJJJJFFJ-FF<FFJJJJJJ7AFFF<FJJ-<-F7<7<A-AF7AJAJ-A-7FF7JJJFJJFJJFFFFAJFJJ7-7-A-F--777FJFJF7---AAAF<AAA--))--))-<))-------)))7- NM:i:2 MD:Z:0A58C15 MQ:i:0 AS:i:69 XS:i:29 SA:Z:chr8,43877720,+,73S54M23S,44,1;
SRR13849430.2 2121 chr8 43877720 44 73H54M23H = 43877720 0 AATCCGCACATCACAAAGCCATTTCACAGACAGCTTCTTTCTACTTTTTATCTG A-7FF7JJJFJJFJJFFFFAJFJJ7-7-A-F--777FJFJF7---AAAF<AAA- NM:i:1 MD:Z:5A48 MQ:i:0 AS:i:49 XS:i:35 SA:Z:chr8,43879758,+,75M75S,60,2;
Contacts extraction
Challenges of contacts extraction solved by pairtools:
multiple walks in each Hi-C read
alignments spaced by short genomic distance may be reported as single alignment with a gap by bwa
PCR and optical duplicates
pairtools parse
The next step is to parse .sam/.bam into pairs. pairtools parse automatically detect the file format.
Full parse documentation: https://github.com/open2c/pairtools/blob/master/doc/parsing.rst
[ ]:
%%bash
pairtools parse -o test.pairs.gz -c ~/.local/share/genomes/hg38/hg38.fa.sizes \
--drop-sam --drop-seq --output-stats test.stats \
--assembly hg38 --no-flip \
--add-columns mapq \
--walks-policy mask \
test.bam
Parse will result in .pairs file. .pairs is a simple tabular format for storing DNA contacts detected in a Hi-C experiment. The detailed .pairs specification is defined by the 4DN Consortium.
[3]:
%%bash
# Header
gzip -dc test.pairs.gz | head -n 5
## pairs format v1.0.0
#shape: whole matrix
#genome_assembly: hg38
#chromsize: chr1 248956422
#chromsize: chr10 133797422
[4]:
%%bash
# Last lines of the header contain the list of columns:
gzip -dc test.pairs.gz | grep "#" | tail -n 5
#samheader: @SQ SN:chrY_KI270740v1_random LN:37240
#samheader: @HD VN:1.5 SO:unsorted GO:query
#samheader: @PG ID:bwa PN:bwa VN:0.7.18-r1243-dirty CL:bwa mem -t 5 -SP /home/agalicina/.local/share/genomes/hg38/index/bwa/hg38.fa SRR13849430_1.fastq.gz SRR13849430_2.fastq.gz
#samheader: @PG ID:pairtools_parse PN:pairtools_parse CL:/home/agalicina/anaconda3/envs/test/bin/pairtools parse -o test.pairs.gz -c /home/agalicina/.local/share/genomes/hg38/hg38.fa.sizes --drop-sam --drop-seq --output-stats test.stats --assembly hg38 --no-flip --add-columns mapq --walks-policy mask test.bam PP:bwa VN:1.1.3
#columns: readID chrom1 pos1 chrom2 pos2 strand1 strand2 pair_type mapq1 mapq2
[5]:
%%bash
# Body of the .pairs file
gzip -dc test.pairs.gz | grep -v "#" | head -n 5
SRR13849430.1 chr12 78795816 ! 0 - - UN 60 0
SRR13849430.2 ! 0 ! 0 - - WW 60 0
SRR13849430.3 chr2 72005391 ! 0 + - UN 60 0
SRR13849430.4 chr2 20530788 ! 0 + - UN 60 0
SRR13849430.5 ! 0 ! 0 - - WW 0 0
pairtools sort
After parsing each pair is listed in the orderthat it appeared in the reads file. Sorting makes it more organised, and also helps to perform deduplication which cannot work on non-sorted files.
[ ]:
%%bash
pairtools sort --nproc 5 -o test.sorted.pairs.gz test.pairs.gz
pairtools dedup
[ ]:
%%bash
pairtools dedup \
--max-mismatch 3 \
--mark-dups \
--output \
>( pairtools split \
--output-pairs test.nodups.pairs.gz \
--output-sam test.nodups.bam \
) \
--output-unmapped \
>( pairtools split \
--output-pairs test.unmapped.pairs.gz \
--output-sam test.unmapped.bam \
) \
--output-dups \
>( pairtools split \
--output-pairs test.dups.pairs.gz \
--output-sam test.dups.bam \
) \
--output-stats test.dedup.stats \
test.sorted.pairs.gz
[6]:
%%bash
# Unique pairs:
gzip -dc test.nodups.pairs.gz | grep -v "#" | head -n 10
SRR13849430.4544778 chr1 136880 chr1 134881 + + UR 28 3
SRR13849430.4112679 chr1 136992 chr1 136650 - + UU 17 12
SRR13849430.1113517 chr1 599102 chr1 599228 + - UU 22 22
SRR13849430.1262300 chr1 656693 chr1 886234 + - UU 5 60
SRR13849430.2778343 chr1 760774 chr1 808155 - + UU 6 21
SRR13849430.3231567 chr1 777267 chr1 35948382 + - UU 32 60
SRR13849430.4836373 chr1 778257 chr1 777890 - + UU 8 48
SRR13849430.1256119 chr1 785664 chr1 954301 + + UU 60 60
SRR13849430.2390636 chr1 788677 chr1 22439216 - + UU 8 60
SRR13849430.3622660 chr1 788734 chr1 822206 + - RU 60 26
[7]:
%%bash
# Only duplicated pairs:
gzip -dc test.dups.pairs.gz | grep -v "#" | head -n 10
SRR13849430.2781007 chr1 760774 chr1 808155 - + DD 6 20
SRR13849430.1103881 chr1 855594 chr1 856474 + - DD 42 47
SRR13849430.3637378 chr1 860367 chr1 863893 - - DD 50 42
SRR13849430.3708228 chr1 860367 chr1 863893 - - DD 50 43
SRR13849430.1681017 chr1 904370 chr1 952493 - - DD 60 60
SRR13849430.4712708 chr1 912376 chr1 913168 - - DD 60 60
SRR13849430.169202 chr1 920729 chr1 929007 - + DD 60 60
SRR13849430.1735365 chr1 929632 chr1 925420 + + DD 60 60
SRR13849430.1743119 chr1 929632 chr1 925420 + + DD 60 60
SRR13849430.1755818 chr1 929632 chr1 925420 + + DD 60 60
pairtools select
Sometimes you may need certain types of pairs based on their properties, such as mapq, pair type, distance or orientation. For all these manipulations, there is pairtools select which requires a file and pythonic condition as an input:
[ ]:
%%bash
pairtools select "mapq1>0 and mapq2>0" test.nodups.pairs.gz -o test.nodups.UU.pairs.gz
pairtools stats
Describe the types fo distance properties of pairs:
[ ]:
%%bash
pairtools stats test.pairs.gz -o test.stats
MultiQC
To make a nice HTML with pairs stats, install Open2C version of multiQC from: https://github.com/open2c/MultiQC
[ ]:
%%bash
multiqc test.stats
[10]:
from IPython.display import IFrame
IFrame(src='./multiqc_report.html', width=1200, height=700)
Load pairs to cooler
Finally, when you obtained a list of appropriate pairs, you may create coolers with it:
[ ]:
%%bash
cooler cload pairs \
-c1 2 -p1 3 -c2 4 -p2 5 \
--assembly hg38 \
~/.local/share/genomes/hg38/hg38.fa.sizes:1000000 \
test.nodups.UU.pairs.gz \
test.hg38.1000000.cool
[ ]:
%%bash
cooler zoomify \
--nproc 5 \
--out test.hg38.1000000.mcool \
--resolutions 1000000,2000000 \
--balance \
test.hg38.1000000.cool
Visualize cooler
Based on open2c vis example
[11]:
import cooler
import matplotlib as mpl
import matplotlib.pyplot as plt
%matplotlib inline
import cooltools.lib.plotting
from matplotlib.colors import LogNorm
import seaborn as sns
import bioframe
import numpy as np
[12]:
file = "test.hg38.1000000.mcool::/resolutions/1000000"
[13]:
clr = cooler.Cooler(file)
[14]:
# Define chromosome starts
chromstarts = []
for i in clr.chromnames:
chromstarts.append(clr.extent(i)[0])
[15]:
from matplotlib.ticker import EngFormatter
bp_formatter = EngFormatter('b')
def format_ticks(ax, x=True, y=True, rotate=True):
if y:
ax.yaxis.set_major_formatter(bp_formatter)
if x:
ax.xaxis.set_major_formatter(bp_formatter)
ax.xaxis.tick_bottom()
if rotate:
ax.tick_params(axis='x',rotation=45)
[20]:
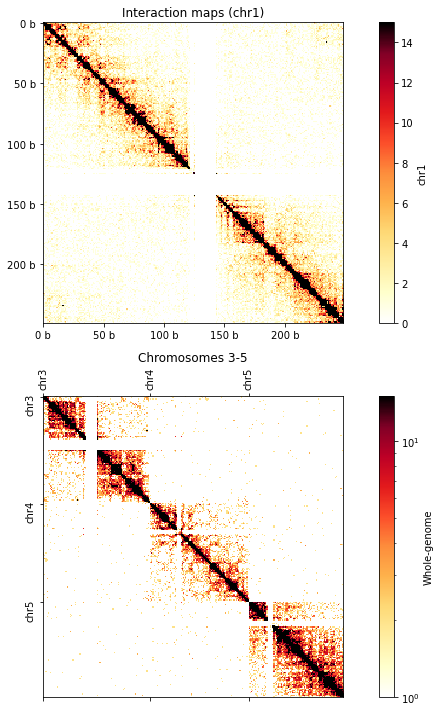
# Define the bounds of the continuous fragment of whole-genome interaction map
chrom_start, chrom_end = clr.chromnames.index('chr10'), clr.chromnames.index('chr11')
start, end = chromstarts[chrom_start], chromstarts[chrom_end]
[21]:
vmax = 15
norm = LogNorm(vmin=1, vmax=vmax)
f, axs = plt.subplots(
figsize=(13, 10),
nrows=2,
ncols=1,
sharex=False, sharey=False)
ax = axs[0]
ax.set_title('Interaction maps (chr1)')
im = ax.matshow(clr.matrix(balance=False).fetch('chr1'), vmax=vmax, cmap='fall');
plt.colorbar(im, ax=ax ,fraction=0.046, pad=0.04, label='chr1');
ax = axs[1]
ax.set_title('Chromosomes 10-11')
im = ax.matshow(clr.matrix(balance=False)[start:end, start:end], norm=norm, cmap='fall');
plt.colorbar(im, ax=ax ,fraction=0.046, pad=0.04, label='Whole-genome');
ax.set_xticks(np.array(chromstarts[chrom_start:chrom_end])-start, clr.chromnames[chrom_start:chrom_end], rotation=90);
ax.set_yticks(np.array(chromstarts[chrom_start:chrom_end])-start, clr.chromnames[chrom_start:chrom_end], rotation=90);
format_ticks(axs[0], rotate=False)
plt.tight_layout()
[ ]: