Overview
pairtools is a simple and fast command-line framework to process sequencing data from a Hi-C experiment. pairtools perform various operations on Hi-C pairs and occupy the middle position in a typical Hi-C data processing pipeline:
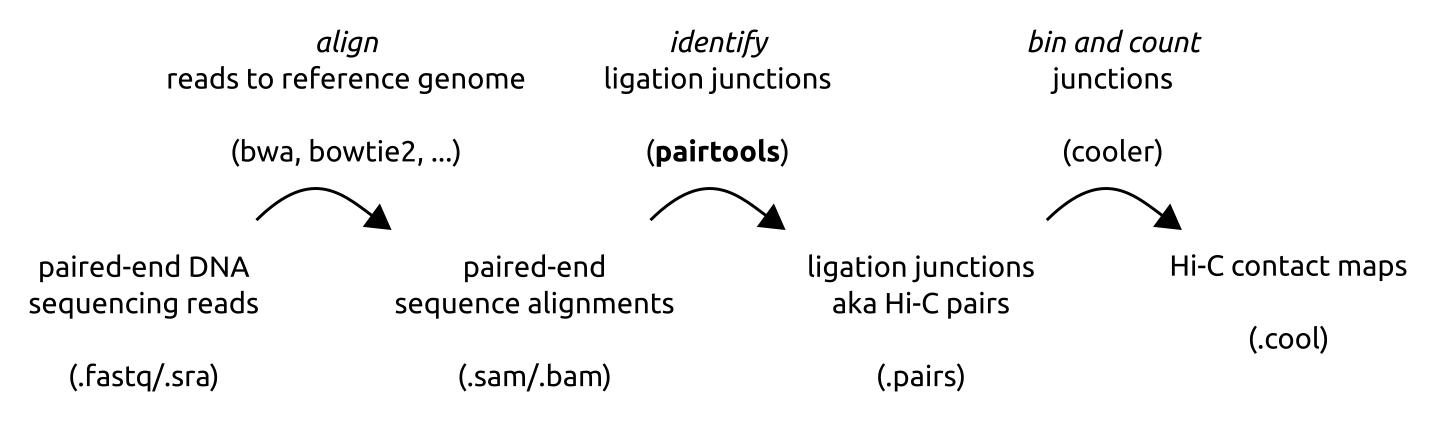
In a typical Hi-C pipeline, DNA sequences (reads) are aligned to the reference genome, converted into ligation junctions and binned, thus producing a Hi-C contact map.
pairtools aim to be an all-in-one tool for processing Hi-C pairs, and can perform following operations:
detect ligation junctions (a.k.a. Hi-C pairs) in aligned paired-end sequences of Hi-C DNA molecules
sort .pairs files for downstream analyses
detect, tag and remove PCR/optical duplicates
generate extensive statistics of Hi-C datasets
select Hi-C pairs given flexibly defined criteria
restore .sam alignments from Hi-C pairs
pairtools produce .pairs files compliant with the 4DN standard.
pairtools uses a two-character notation to define pair types (see table _section-pair-types)
The full list of available pairtools:
Pairtool |
Description |
|---|---|
dedup |
Find and remove PCR/optical duplicates. |
filterbycov |
Remove pairs from regions of high coverage. |
flip |
Flip pairs to get an upper-triangular matrix. |
markasdup |
Tag pairs as duplicates. |
merge |
Merge sorted .pairs/.pairsam files. |
parse |
Find ligation junctions in .sam, make .pairs. |
phase |
Phase pairs mapped to a diploid genome. |
restrict |
Assign restriction fragments to pairs. |
select |
Select pairs according to some condition. |
sort |
Sort a .pairs/.pairsam file. |
split |
Split a .pairsam file into .pairs and .sam. |
stats |
Calculate pairs statistics. |
Contents:
Tutorials